Dystrophies rétiniennes héréditaires
Revu par Dr Philippa Vincent, MRCGPDernière mise à jour par Dr Colin Tidy, MRCGPLast updated 21 juil. 2023
Respecte les directives éditoriales
- TéléchargerTélécharger
- Partager
- Language
- Discussion
- Version audio
- Add to preferred sources on Google
Professionnels de la santé
Professional Reference articles are designed for health professionals to use. They are written by UK doctors and based on research evidence, UK and European Guidelines. You may find the Problèmes visuels article more useful, or one of our other articles de santé.
Dans cet article:
Continuez à lire ci-dessous
Aperçu1
Hereditary retinal dystrophies are a large group of inherited eye disorders resulting in irreversible visual loss. They develop due to mutations in one or more genes that lead to the death of the retinal photoreceptor cells. Mutations in over 200 genes are known to be associated with all different forms of retinal disorders.
Therapeutic avenues that are being investigated for these disorders include gene therapy to replace the defective gene, treatment with neurotrophic factors to stimulate the growth of photoreceptors, cell replacement therapy, and prosthetic devices that can capture light and transmit electrical signals through retinal neurons to the brain. A combination of approaches that involve both gene replacement and cell replacement may be required for optimum benefit.
Retinitis pigmentosa is the most common retinal dystrophy affecting 1 in 3,000 individuals. Most retinitis pigmentosa mutations affect rods selectively. See separate Retinitis Pigmentosa article.
By contrast with retinitis pigmentosa, in which degeneration of the rods initially affects the peripheral retina, many inherited retinal dystrophies primarily affect the macula. Macular dystrophies are characterised by gradual loss of acuity, colour vision and contrast sensitivity with onset usually by the second decade of life. There is a variety of (often overlapping) names.
Correct diagnosis is important, as it determines the expected prognosis. Specific gene therapy for a number of diseases is being evaluated.2
Anatomy of the retina
Retour au sommaireThe retina is a many layered structure and retinal dystrophies may affect any of the layers. From the vitreous to the choroid these are the internal limiting membrane, nerve fibre layer, ganglion cell layer, inner plexiform layer, inner nuclear layer, outer plexiform layer, outer nuclear layer, external limiting membrane, rod and cone inner and outer segments and the retinal pigment epithelium (RPE).
The macula has a high density of cones, ganglion cells, and pigment within bipolar and ganglion cells. The central 1.5 mm area of the macula is the fovea. Within the fovea is a roughly circular avascular area, the foveal avascular zone, which contains only cones.
Anatomie de la rétine
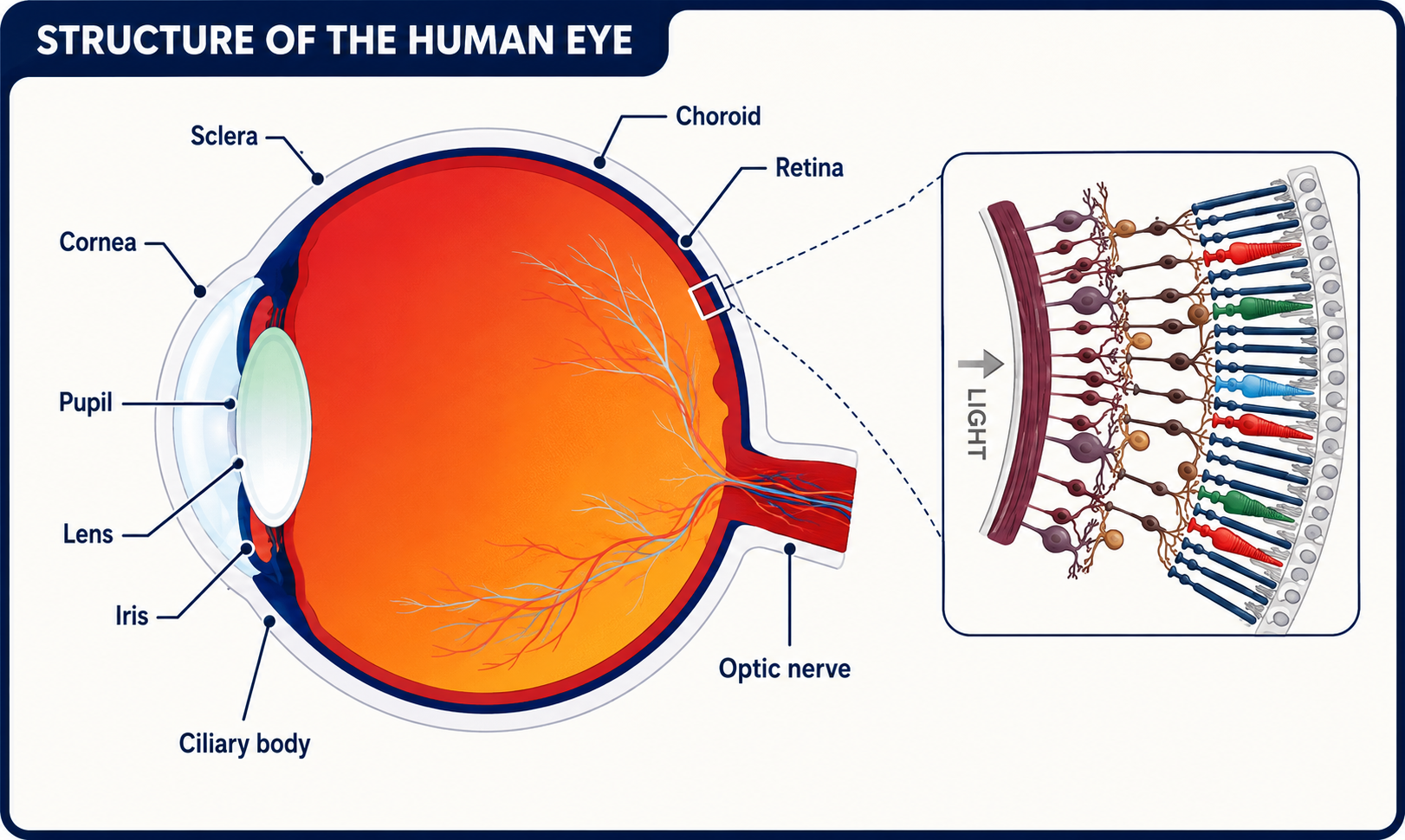
© د.مصطفى الجزار, CC BY-SA 3.0, via Wikimedia Commons
Continuez à lire ci-dessous
Hereditary retinal dystrophies symptoms (presentation)2
Retour au sommaireThis varies by disease. Within the retina there are 60-125 million rods and 3.2-6.5 million cones. No rods are present in the fovea, although the highest density of rods is found at a distance of about 20° from the fovea. The cones are mainly concentrated in the fovea and are most densely packed within the foveola. Therefore, diseases affecting rods tend to cause visual problems at night as well as peripheral visual field defects. Diseases affecting cones cause increased sensitivity to light, loss of central vision, impaired colour vision and central visual field defects.
Diagnostic2
Retour au sommaireDiagnosis is generally made on clinical examination and subjective testing, although it needs confirmation with electro-diagnostic tests. This helps differentiate retinal disease from choroidal disease and helps ensure accuracy of diagnosis (important in view of the genetic implications).
Fundoscopy - also using red-free light - visual field tests, electrophysiological tests and multifocal electroretinograms (ERGs) and tests evaluating colour vision may all assist in the diagnosis.
Subjective retinal tests
Colour vision tests: the best known of these are the Ishihara's test plates which distinguish between red/green colour blindness. Other, more sophisticated tests can assess for yellow colour blindness, as well as help diagnose complex and subtle degrees of colour blindness.
Dark adaptometry is useful in patients complaining of night blindness (nyctalopia) - which is commonly a feature of these disorders.
Objective retinal assessment
Fluorescein angiography may help differentiate retinal from choroidal disease.
ERGs record the action potential produced by the retina in response to light and show typical patterns in dark (scotopic) and light (photopic) conditions. Deviations from normal waves assist diagnosis in a similar manner to an ECG. During the test, an electrode is placed on the cornea to measure the retina's electrical responses to light.
Electro-oculograms (EOGs) complement ERG measurements. They measure the standing potential between the electrically positive cornea and the electrically negative back of the eye. An abnormal EOG arises as a result of problems in the RPG.
New methods are being evaluated, such as high-resolution spectral-domain optic coherence tomography (SD-OCT) and fundus autofluorescence.
Tests génétiques2
Conventional genetic testing would be expensive and time-consuming, as mutations in more than 200 genes are involved across the diverse range of dystrophies. New methods of genetic testing offer the possibility of analysing multiple genes. The methods allow much faster and more cost-effective analyses.
Continuez à lire ci-dessous
Hereditary retinal dystrophies treatment and management 2 3
Retour au sommaireManagement is focused on diagnosis and specialised genetic counselling. Treatment options are limited and tend to be concentrated around optometric visual rehabilitation (eg, use of aids for low vision, orientation and mobility training). Work is also underway in the field of molecular and gene therapy, as the responsible genes are increasingly being identified.
The availability and provision of the highly specialised multidisciplinary services patients need vary throughout the UK.
There is promising research in the area of gene therapy - for example, in Leber's congenital amaurosis, which is associated with dysfunction and degeneration of photoreceptors, studies have shown some positive effects of gene therapy.
Examples of hereditary retinal disorders4
Retour au sommaireThere are a variety of hereditary retinal dystrophies, some of which are very rare. The hallmark is a loss of visual acuity. These conditions are a major cause of severe sight impairment and affect patients at all ages. They vary at the genetic level, as well as at the clinical, histological and physiopathological levels. Ongoing genetic research continues to change the understanding of pathophysiology.
Stargardt's disease and fundus flavimaculatus2
There has been some question as to whether this condition is two diseases or one. Stargardt's disease or fundus flavimaculatus is a progressive form of juvenile macular degeneration with clinical and genetic heterogeneity. Mutations in at least four genes are responsible for similar clinical characteristics and there is a lack of clear diagnostic distinction between them. Stargardt's disease and fundus flavimaculatus are discussed here as one entity.
Stargardt's disease, with or without fundus flavimaculatus, is the most common hereditary dystrophy affecting the central retina, occurring in 1 in 8,000-10,000 people.5
Description - also known as juvenile macular dystrophy: this is one of the two most common forms of inherited macular degeneration. It accounts for 7% of all retinal dystrophies. First described by the German ophthalmologist Karl Stargardt in 1909, it is a progressive, bilateral atrophic macular dystrophy characterised by perimacular and peripheral 'dirty grey-yellow spots' (fundus flavimaculatus).
Hérédité - mainly autosomal-recessive; there is a rare autosomal-dominant variant.
Présentation - childhood (aged about 6 years) to early adulthood: bilateral (usually) decreased central vision. This is often out of proportion to the clinical picture. There is also progressive colour blindness.
Pronostic - generally poor. Most patients experience rapid deterioration of vision during the first two decades of life. Once vision drops below 6/12, progression is rapid and the visual prognosis is poor. Visual rehabilitation can achieve and maintain some degree of independence.6
A variant which has been named fundus flavimaculatus presents later and the macula may be spared. The patient may present with central visual deterioration. Deterioration in colour vision may not be noticed until later. The distribution and number of yellowish spots may change over the course of time. Prognosis tends to be better and peripheral and night vision are unaffected.
Juvenile Best's disease (Best's vitelliform macular dystrophy)2
Best's disease was identified by Friedrich Best in 1906 and is the second most common hereditary dystrophy of the macula.
Description - it is characterised by an abnormal accumulation of lipofuscin at the level of the RPG. This grows over years, eventually to give rise to a characteristic round egg-yolk appearance and which may be later associated with a pseudo-hypopyon.
Hérédité - It is a progressive autosomal-dominant hereditary disease with variable penetrance among different members of the same family. The mutation has been localised to the BEST1 (VMD2) gene on chromosome 11.
Présentation - symptoms may start within the first two decades of life. Visual acuity deteriorates when the 'egg-yolk lesion' ruptures. Changes occur in EOG readings in children before they are symptomatic. Vision may be only slightly decreased in childhood and teenage years when the 'egg-yolk lesion' is present. Typically vision is not significantly affected until the fifth decade.
Pronostic - declining visual acuity may be a reflection of macular scarring but other complications can add to this (eg, macular hole formation or décollement de la rétine). Some patients go on to become legally severely sight impaired but most individuals retain reading ability with vision in one eye.
Adult vitelliform foveomacular dystrophy (adult vitelliform degeneration)2 7
Description - in this disease, there are bilateral, symmetrical lesions within the macula. They are similar to those of Best's disease but they are smaller, they present in adulthood and they do not progress.
Hérédité - probably autosomal-dominant.
Présentation - aged 40-50 years: blurred vision ± distortion of images (metamorphopsia) which may be mild to the point that this condition is often discovered by chance.
Pronostic - good, unless complications occur (such as neovascularisation of the underlying choroid).
Other hereditary macular dystrophies4
Sorsby's pseudo-inflammatory macular dystrophy, North Carolina macular dystrophy and dominant cystoid macular oedema carry poor prognoses.
Butterfly macular dystrophy is a relatively innocuous condition (often found by chance) resulting in mild impairment of central vision.
Pattern dystrophy presents with paracentral distortion and loss of visual acuity. It may also be asymptomatic. Little yellow flecks are seen throughout the fundus, with associated macular atrophy.
Achromatopsia8
Description - this condition causes complete loss of cone function, while rod function is normal throughout the course of disease. Its prevalence is estimated to be about 1:30,000. Since cones are concentrated at the fovea then the macula and fovea are disproportionately affected.
Hérédité - congenital achromatopsia is transmitted in an autosomal-recessive trait
Présentation - patients present in early childhood with nystagmus, abnormal visual behaviour or photophobia. Acuity is less than 20/200. Fundoscopy is unremarkable. Visual fields show a relative central scotoma. ERG is the most useful tool in assessing function. It shows that cone function is missing while rod function is normal. Mutations in several genes have been detected which cause achromatopsia.
Pronostic - there is no progression and no treatment other than low vision support. Gene therapy has shown early promise: restoration of cone function has been attained in animal models with similarities to the human phenotype, using virally mediated gene replacement therapy with different forms of a red cone opsin promoter .
X-linked retinoschisis2
Description and inheritance - this is an X-linked recessive disease, caused by mutations of the RS1 gene, which encodes the protein retinoschisin. This results in retinoschisis, or splitting of the retina's layers, usually in the outer plexiform layer. The affected part of the retina will have suboptimal vision.
Présentation - it almost exclusively affects young males. The prevalence is estimated to be about 1:15,000-1:30,000. Peripheral retinoschisis is seen in about half of patients. Foveal retinoschisis is present in almost all patients and can be seen with SD-OCT which shows cystic spaces within the retina. Central vision can be impaired, with visual acuity ranging from 20/30 to less than 20/200. Acuity loss is caused by the formation of tiny cysts between the separated layers of the retina. These cysts often form a 'spoke-wheel' pattern. Peripheral vision can also be lost if the inner layer of nerve cells splits off from the outer layer of cells.
Pronostic - recently, the topical administration of carbonic anhydrase inhibitors dorzolamide and brinzolamide was shown to be effective in reducing the macular cysts in patients with X-linked retinoschisis.
Familial drusen (North Carolina macular dystrophy)9
Description - this condition (with subtypes known as Doyne's honeycomb choroiditis or malattia leventinese) is thought to be an early form of age-related macular degeneration. It is characterised by well-defined creamy-yellow spots (drusen) over the macula, which can eventually extend widely over the posterior pole of the eye and around the optic disc.
Hérédité - autosomal-dominant with full penetrance but variable expressivity.
Présentation - the drusen only tend to become visually problematic around the fifth decade of life and patients then experience central visual decline.
Pronostic - this is a progressive disease but peripheral vision is spared.
Bietti's crystalline dystrophy10
Description - this condition is characterised by crystalline depositions in the peripheral cornea and the retina. This may be accompanied by progressive retinal atrophy.
Hérédité - X-linked or autosomal-recessive.
Présentation - there is progressive visual loss in the third decade, particularly peripheral and night vision.
Pronostic - the rate of progression varies between individuals.
Progressive cone dystrophies11
Description - this group of rare disorders encompasses a range of problems from pure cone dysfunction to those with varying degrees of associated (but usually less severe) rod dysfunction. Many patients start with a pure cone problem which then progressively affects the rods over time. Their estimated incidence ranges from 1 in 20,000–100,000.
Hérédité - most are sporadic but autosomal-dominant, autosomal-recessive and X-linked inheritance is also known.
Présentation - slow, progressive, bilateral visual loss (night vision better than day), photophobia, poor colour vision ± nystagmus. There may also be associated visual field defects.
Pronostic - short-term, those with less rod involvement do well but, ultimately, the outlook is poor.
Syndrome d'Alport12
Alport's syndrome is a genetic disorder affecting around 1 in 5,000 children, causing glomerulonephritis, end-stage kidney disease and sensorineural hearing loss. It can affect the eyes.
Description - the condition affects collagen type IV synthesis and results in basement membrane abnormalities which manifest themselves through chronic kidney disease and sometimes with sensorineural deafness. Various eye abnormalities are often seen including lenticonus (cone-shaped lens), keratoconus, cataracts and retinal flecks in the macula and mid-periphery. These rarely threaten vision.
Hérédité - X-linked dominant.
Présentation - with renal problems and, although not invariably, sensorineural deafness.
Pronostic - the visual prognosis is good; acuity is not normally affected. Lenticonus can be treated by replacement of the lens, as for cataracts. Mild keratoconus can be treated with hard or piggy-back contact lenses; severe cases may require a corneal transplant.
Benign familial fleck retina13 14
In this rare condition the fundi are massively invaded by lesions which appeared as discrete, bright white or yellow flecks situated well behind the retinal blood vessels. The macula is always spared. Fluorescein studies reveal a healthy macula and retinal and choroidal blood vessels.
Description - benign fovea-sparing irregularly shaped lesions which densely cover the retina, extending to the far periphery. They may appear white, yellow or grey.
Hérédité - autosomal-recessive.
Présentation - asymptomatic, usually discovered by chance.
Pronostic - excellent, vision is not normally affected.
Leber's congenital amaurosis (LCA)15 16 17
LCA is the severest of all retinal dystrophies. Affected individuals usually present in the first year of life with profound sight impairment, roving nystagmus, variable retinal pathology and occasionally other systemic pathology. It is now known to be caused by at least six genes. Different mutations in several of these genes have been observed to cause retinitis pigmentosa and other retinal dystrophies. It is suggested that the LCA phenotype must therefore result from mutations which have the severest consequence.
The estimated prevalence is 1 : 50,000-100,000 - it is an early-onset inherited cause of severe sight impairment in childhood, characterised by a severe retinal dystrophy. It typically presents in the first year of life.
Description - visual function is usually poor and often accompanied by nystagmus, sluggish or absent pupillary responses, photophobia, high hyperopia and keratoconus. Visual acuity is rarely better than 20/400.
Hérédité - variants in at least six genes have been associated with LCA or early-onset retinitis pigmentosa, leading to genetically heterogeneous inheritance.
Présentation - children are severely sight impaired either from birth or within the first few years of life. A characteristic finding is 'Franceschetti's oculo-digital sign', comprising eye poking, pressing and rubbing. Constant rubbing of the eyes results in orbital fat resorption and subsequent endophthalmos (eyes sunken into sockets). The retina may initially appear normal but a pigmentary retinopathy similar to retinitis pigmentosa is frequently observed later in childhood. The ERG is characteristically non-detectable or severely subnormal.
Pronostic - treatment is supportive. Children benefit from correction of refractive error, use of aids for low vision when possible and support in educational opportunities. When possible, children should be discouraged from repeatedly poking and pressing on their eyes. In those with residual vision, assessment for amblyopia, glaucoma or cataract is needed regularly. Gene therapy may offer benefit in the future: trials of gene replacement therapy have proved successful in restoring sight to such affected dogs.18
Congenital stationary night blindness16 19
This is a group of rare, non-progressive conditions of the retina in which abnormal rod function causes impaired night vision. Most familial cases are X-linked, although autosomal dominance has been described. There are associated ocular symptoms such as myopia, hyperopia, nystagmus and reduced visual acuity.
Description - the prevalence of this condition is unknown but very low. It is present from birth.
Hérédité - depending on the subtype, this may be autosomal-dominant, or autosomal-recessive, or X-linked.
Présentation - poor night vision as a result in a delay or an inability to achieve normal dark-adapted rod thresholds. Patients have night blindness, reduced acuity, high myopia, nystagmus and strabismus. There are two major types - the complete form and the incomplete form. The complete form always causes night blindness, while the incomplete form does not always do so. The types are distinguished by ERG.
Pronostic - there is no progression.
Congenital monochromatism20
Description - this is an umbrella term used to describe various degrees of rod monochromatism or cone monochromatism. It can be associated with disorders of endocrine or hypothalamic function and with sensorineural deafness.
Hérédité - depending on subtype, autosomal-recessive or X-linked.
Présentation - colour blindness: if colour vision is totally absent, the world is seen in shades of grey. There may be some colour perception in incomplete rod monochromatism. Cone monochromatism is associated with better visual acuity (6/6 to 6/9) than rod monochromatism (where this is complete, visual acuity is in the region of 6/60).
Pronostic - there is no progression.
Dr Mary Lowth est l'auteur ou l'auteur original de ce dépliant.
Exclusive updates for healthcare professionals
Stay informed with the latest clinical updates, professional insights, and evidence-based guidance. The Patient Pro newsletter curates essential content for healthcare professionals—delivered straight to your inbox.
By subscribing you accept our Politique de confidentialité. Vous pouvez vous désabonner à tout moment. Nous ne vendons jamais vos données.
Lectures complémentaires et références
- Institut National Royal des Personnes Aveugles (RNIB)
- Leber's Congenital Amaurosis; Contact a Family
- Ziccardi L, Cordeddu V, Gaddini L, et al; Gene Therapy in Retinal Dystrophies. Int J Mol Sci. 2019 Nov 14;20(22):5722. doi: 10.3390/ijms20225722.
- Solebo AL, Teoh L, Rahi J; Epidemiology of blindness in children. Arch Dis Child. 2017 Sep;102(9):853-857. doi: 10.1136/archdischild-2016-310532. Epub 2017 May 2.
- Brito-Garcia N, Del Pino-Sedeno T, Trujillo-Martin MM, et al; Effectiveness and safety of nutritional supplements in the treatment of hereditary retinal dystrophies: a systematic review. Eye (Lond). 2017 Feb;31(2):273-285. doi: 10.1038/eye.2016.286. Epub 2016 Dec 9.
- Kannabiran C, Mariappan I; Therapeutic avenues for hereditary forms of retinal blindness. J Genet. 2018 Mar;97(1):341-352.
- Nentwich MM, Rudolph G; Hereditary retinal eye diseases in childhood and youth affecting the central retina. Oman J Ophthalmol. 2013 Sep;6(Suppl 1):S18-25. doi: 10.4103/0974-620X.122290.
- Moore T, Burton H; Genetic Ophthalmology in Focus: A Needs Assessment & Review of Specialist Services for Genetic Eye Disorders, April 2008.
- Rozet JM, Gerber S, Ducroq D, et al; Hereditary macular dystrophies. J Fr Ophtalmol. 2005 Jan;28(1):113-24.
- Thumann G; Prospectives for gene therapy of retinal degenerations. Curr Genomics. 2012 Aug;13(5):350-62. doi: 10.2174/138920212801619214.
- Shah M, Zaman M, Khan MT, et al; Visual rehabilitation of patients with Stargardt's disease. J Coll Physicians Surg Pak. 2008 May;18(5):294-8.
- Do P, Ferrucci S; Adult-onset foveomacular vitelliform dystrophy. Optometry 2006 Apr;77(4):156-66.
- Komaromy AM, Alexander JJ, Rowlan JS, et al; Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet. 2010 Jul 1;19(13):2581-93. doi: 10.1093/hmg/ddq136. Epub 2010 Apr 8.
- Zhu Z, Zou H, Li C, et al; The possible pathogenesis of macular caldera in patients with North Carolina macular dystrophy. BMC Ophthalmol. 2022 Nov 19;22(1):447. doi: 10.1186/s12886-022-02655-w.
- Vargas M, Mitchell A, Yang P, et al; Bietti Crystalline Dystrophy. GeneReviews®, April 2012.
- Gill JS, Georgiou M, Kalitzeos A, et al; Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy. Br J Ophthalmol. 2019 Jan 24;103(5):711-20. doi: 10.1136/bjophthalmol-2018-313278.
- Savige J, Lipska-Zietkiewicz BS, Watson E, et al; Guidelines for Genetic Testing and Management of Alport Syndrome. Clin J Am Soc Nephrol. 2022 Jan;17(1):143-154. doi: 10.2215/CJN.04230321. Epub 2021 Dec 20.
- Bin NJ, Heng HM, Poh R, et al; Phospholipase A2 group v in benign familial fleck retina in a set of triplets. Retina. 2015 Jun;35(6):1266-72. doi: 10.1097/IAE.0000000000000446.
- Sabel Aish SF, Dajani B; Benign familial fleck retina. Br J Ophthalmol. 1980 Sep;64(9):652-9.
- Francis PJ; Genetics of inherited retinal disease. J R Soc Med. 2006 Apr;99(4):189-91.
- Pusch CM, Zeitz C, Brandau O, et al; The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat Genet. 2000 Nov;26(3):324-7.
- Weleber RG, Francis PJ, Trzupek KM, et al; Leber Congenital Amaurosis
- Drack AV, Johnston R, Stone EM; Which Leber congenital amaurosis patients are eligible for gene therapy trials? J AAPOS. 2009 Oct;13(5):463-5.
- Abramowicz MJ, Ribai P, Cordonnier M; Congenital stationary night blindness: report of an autosomal recessive family and linkage analysis. Am J Med Genet A. 2005 Jan 1;132A(1):76-9.
- Jan JE, Tze WJ, Johnston AC, et al; Familial congenital monochromatism, cataracts, and sensorineural deafness. Am J Dis Child. 1976 Dec;130(12):1349-50.
Continuez à lire ci-dessous
About the authorView full bio

Dr Colin Tidy, MRCGP
Médecin généraliste, Auteur médical
MBBS, MRCGP, MRCP (Paediatrics), DCH
Dr Colin Tidy is an NHS Doctor, based in Oxfordshire.
About the reviewerView full bio

Dr Philippa Vincent, MRCGP
Médecin généraliste, Auteur médical
MB BS, Bsc, MRCGP (2000), DCH, DFSRH, DRCOG
Dr Philippa Vincent is an NHS GP working in North London.
Historique de l'article
Les informations sur cette page sont rédigées et examinées par des cliniciens qualifiés.
Prochaine révision prévue : 19 juil. 2028
21 juil. 2023 | Dernière version

Demandez, partagez, connectez-vous.
Parcourez les discussions, posez des questions et partagez vos expériences sur des centaines de sujets de santé.

Vous ne vous sentez pas bien ?
Évaluez vos symptômes en ligne gratuitement